The first Bay Area Chromatin and Epigenomics group event took place on February 12th 2020 in Menlo Park, CA. It gathered a wide representation of professionals from companies and research institutions across the San Francisco Bay Area.
Cancer genomics
Our first speaker that night was Robert Power – my colleague from Personalis and Global Product Manager in Immuno-oncology, who gave a talk about Personalis’s NeXT platform and the advances it brings to cancer genomics.
After introducing the platform, Robert described a collaborative study that used Personalis’s methods to track the response of patients to immuno-oncology (I-O) treatment with Keytruda (pembrolizumab) or Opdivo (nivolumab), both of which are anti-PD-1 antibodies.
The research was powered by the immunogenomics analysis using ImmunoID NeXT, including tumor mutational burden (TMB), and neoantigen analysis that introduced a new adjusted neoantigen burden metric. The adjusted neoantigen burden appeared to be significantly associated with patients’ progression-free survival and with improved response to the anti-PD-1 therapy.
As the field of immuno-oncology grapples with varied and unpredictable patient outcomes, these results bring hope that one day thanks to immunogenomics we may better understand how to measure and evaluate patient’s responses to immuno-oncology treatments.
Deep learning for epigenomics
Our second speaker was Avantika Lal, Deep Learning and Genomics Scientist from NVIDIA, who talked about her Deep Learning tool, AtacWorks.
Avantika showed us how rapidly the popularity of ATAC-seq has grown in the recent years.
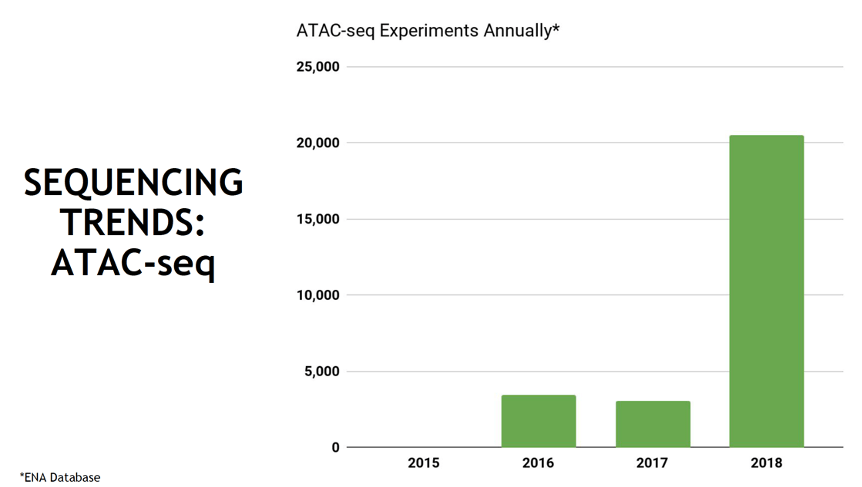
With its relative ease of sample preparation, ATAC-seq quickly became the tool of choice for probing chromatin’s open and closed states, both in bulk and single-cell settings. But low inputs and high expectations can sometimes lead to poor results – and in next generation sequencing (NGS) that means low coverage.
Since ATAC-seq analysis relies on peak-calling based on the pileups of NGS reads mapping to specific genomic regions, low coverage means low certainty of such calls and difficulties with separating the real signal from background noise. This is especially limiting in single-cell ATAC-seq experiments, where the population of interest can often be a very rare subtype of cells mixed in a large population.
That’s where AtacWorks comes to rescue. Trained on paired examples of noisy and clean ATAC-seq datasets, using a deep learning model, AtacWorks is able to de-noise low coverage datasets and enhance accurate peak calling, to the level that allowed the authors to discover new hematopoietic cell states within a noisy single-cell ATAC-seq dataset. The tool, described at BioRxiv and available via github, can save researchers who have to work with scarce material (e.g. low abundance cell population) or difficult material (e.g. old but valuable patient samples).
On to the next one
As the first event ran to its end, my thoughts wandered towards the next one. COVID-19 interfered with those plans, and the second BACE event was postponed.
But what COVID-19 also did was draw our attention to questions of immunity. And so, in April 2020, we held the next BACE event on Epigenomics of Immunity.
